Getting started with tskit#
You’ve run some simulations or inference methods, and you now have a
TreeSequence object; what now? This tutorial is aimed at
users who are new to tskit and would like to get some
basic tasks completed. We’ll look at five fundamental things you might
want to do:
process trees,
process sites & mutations,
extract genotypes,
compute statistics, and
save or export data.
Throughout, we’ll also provide pointers to where you can learn more.
Note
The examples in this tutorial are all written using the Python API, but it’s also possible to use R, or access the API in other languages, notably C and Rust.
A number of different software programs can generate tree sequences. For the purposes of this tutorial we’ll use msprime to create an example tree sequence representing the genetic genealogy of a 10Mb chromosome in twenty diploid individuals. To make it a bit more interesting, we’ll simulate the effects of a selective sweep in the middle of the chromosome, then throw some neutral mutations onto the resulting tree sequence.
import msprime
pop_size=5000
seq_length=10_000_000
sweep_model = msprime.SweepGenicSelection(
position=seq_length/2, start_frequency=0.0001, end_frequency=0.9999, s=0.25, dt=1e-6)
ts = msprime.sim_ancestry(
20,
model=[sweep_model, msprime.StandardCoalescent()],
population_size=pop_size,
sequence_length=seq_length,
recombination_rate=1e-8,
random_seed=1234, # only needed for repeatability
)
# Optionally add finite-site mutations to the ts using the default Jukes & Cantor model, creating SNPs
ts = msprime.sim_mutations(ts, rate=1e-8, random_seed=4321)
ts
|
|
|
|---|---|
| Trees | 5 216 |
| Sequence Length | 10 000 000 |
| Time Units | generations |
| Sample Nodes | 40 |
| Total Size | 1.1 MiB |
| Metadata | No Metadata |
| Table | Rows | Size | Has Metadata |
|---|---|---|---|
| Edges | 16 811 | 525.4 KiB | |
| Individuals | 20 | 584 Bytes | |
| Migrations | 0 | 8 Bytes | |
| Mutations | 6 374 | 230.3 KiB | |
| Nodes | 3 521 | 96.3 KiB | |
| Populations | 1 | 224 Bytes | ✅ |
| Provenances | 2 | 1.9 KiB | |
| Sites | 6 371 | 155.6 KiB |
| Provenance Timestamp | Software Name | Version | Command | Full record |
|---|---|---|---|---|
| 19 June, 2026 at 02:07:05 PM | msprime | 1.4.0 | sim_mutations |
Detailsdictschema_version: 1.0.0
software:
dictname: msprimeversion: 1.4.0
parameters:
dictcommand: sim_mutations
tree_sequence:
dict__constant__: __current_ts__rate: 1e-08 model: None start_time: None end_time: None discrete_genome: None keep: None random_seed: 4321
environment:
dict
os:
dictsystem: Linuxnode: runnervm7b5n9 release: 6.17.0-1018-azure version: #18~24.04.1-Ubuntu SMP Thu May 28 16:39:11 UTC 2026 machine: x86_64
python:
dictimplementation: CPythonversion: 3.11.15
libraries:
dict
kastore:
dictversion: 2.1.2
tskit:
dictversion: 1.0.3
gsl:
dictversion: 2.6 |
| 19 June, 2026 at 02:07:05 PM | msprime | 1.4.0 | sim_ancestry |
Detailsdictschema_version: 1.0.0
software:
dictname: msprimeversion: 1.4.0
parameters:
dictcommand: sim_ancestrysamples: 20 demography: None sequence_length: 10000000 discrete_genome: None recombination_rate: 1e-08 gene_conversion_rate: None gene_conversion_tract_length: None population_size: 5000 ploidy: None
model:
listdictduration: Noneposition: 5000000.0 start_frequency: 0.0001 end_frequency: 0.9999 s: 0.25 dt: 1e-06 __class__: msprime.ancestry.SweepGenicSel ection dictduration: None__class__: msprime.ancestry.StandardCoale scent initial_state: None start_time: None end_time: None record_migrations: None record_full_arg: None additional_nodes: None coalescing_segments_only: None num_labels: None random_seed: 1234 stop_at_local_mrca: None replicate_index: 0
environment:
dict
os:
dictsystem: Linuxnode: runnervm7b5n9 release: 6.17.0-1018-azure version: #18~24.04.1-Ubuntu SMP Thu May 28 16:39:11 UTC 2026 machine: x86_64
python:
dictimplementation: CPythonversion: 3.11.15
libraries:
dict
kastore:
dictversion: 2.1.2
tskit:
dictversion: 1.0.3
gsl:
dictversion: 2.6 |
You can see that there are many thousands of trees in this tree sequence.
Note
Since we simulated the ancestry of 20 diploid individuals, our tree sequence contains 40 sample nodes, one for each genome.
Processing trees#
Moving along a tree sequence usually involves iterating over all of its Tree
objects. This common idiom underlies many tree sequence algorithms, including those
we’ll encounter later in this tutorial for calculating
population genetic statistics.
To iterate over a tree sequence you can use
TreeSequence.trees().
for tree in ts.trees():
print(f"Tree {tree.index} covers {tree.interval}")
if tree.index >= 4:
print("...")
break
print(f"Tree {ts.last().index} covers {ts.last().interval}")
Tree 0 covers Interval(left=0.0, right=613.0)
Tree 1 covers Interval(left=613.0, right=2051.0)
Tree 2 covers Interval(left=2051.0, right=5479.0)
Tree 3 covers Interval(left=5479.0, right=11811.0)
Tree 4 covers Interval(left=11811.0, right=12602.0)
...
Tree 5215 covers Interval(left=9997737.0, right=10000000.0)
In this code snippet, as well as the trees() iterator, we’ve also
used TreeSequence.last() to access the last tree directly; it may not surprise you
that there’s a corresponding TreeSequence.first() method to return the first tree.
Above, we stopped iterating after Tree 4 to limit the printed output, but iterating
forwards through trees in a tree sequence (or indeed backwards using the standard Python
reversed() function) is efficient. That means it’s quick, for example to check if all
the trees in a tree sequence have fully coalesced (which is to be expected in
reverse-time, coalescent simulations, but not always for tree sequences produced by
forward simulation).
import time
elapsed = time.time()
for tree in ts.trees():
if tree.has_multiple_roots:
print(f"Tree {tree.index} has not coalesced")
break
else:
elapsed = time.time() - elapsed
print(f"All {ts.num_trees} trees coalesced")
print(f"Checked in {elapsed:.6g} secs")
All 5216 trees coalesced
Checked in 0.00468564 secs
Now that we know all trees have coalesced, we know that at each position in the genome
all the 40 sample nodes must have one most recent common ancestor (MRCA). Below, we
iterate over the trees, finding the IDs of the root (MRCA) node for each tree. The
time of this root node can be found via the tskit.TreeSequence.node() method, which
returns a Node object whose attributes include the node time:
import matplotlib.pyplot as plt
kb = [0] # Starting genomic position
mrca_time = []
for tree in ts.trees():
kb.append(tree.interval.right/1000) # convert to kb
mrca = ts.node(tree.root) # For msprime tree sequences, the root node is the MRCA
mrca_time.append(mrca.time)
plt.stairs(mrca_time, kb, baseline=None)
plt.xlabel("Genome position (kb)")
plt.ylabel("Time of root (or MRCA) in generations")
plt.yscale("log")
plt.show()
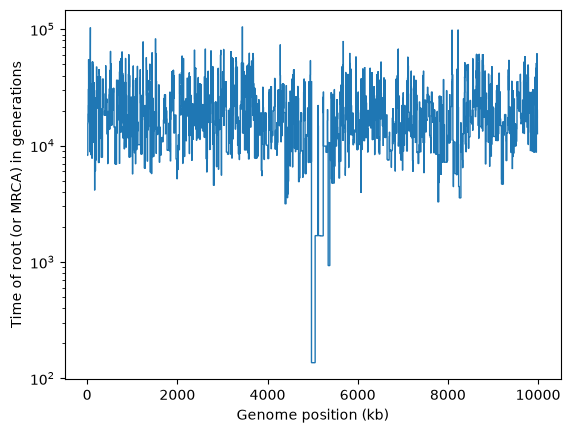
It’s obvious that there’s something unusual about the trees in the middle of this chromosome, where the selective sweep occurred.
Although tskit is designed so that it is rapid to pass through trees sequentially,
it is also possible to pull out individual trees from the middle of a tree sequence
via the TreeSequence.at() method. Here’s how you can use that to extract
the tree at location \(5\ 000\ 000\) — the position of the sweep — and draw it
using the Tree.draw_svg() method.
swept_tree = ts.at(5_000_000) # or you can get e.g. the nth tree using ts.at_index(n)
intvl = swept_tree.interval
swept_tree.draw_svg(
size=(1000, 200), # Draw the tree at a wide size, to make room for all 40 tips
title=f"Tree number {swept_tree.index}, which runs from position {intvl.left} to {intvl.right}:",
y_axis=True,
y_ticks=[0, 20, 40, 60, 80],
node_labels={}
)

This tree shows the classic signature of a recent expansion or selection event, with many long terminal branches, resulting in an excess of singleton mutations.
It can often be helpful to slim down a tree sequence so that it represents the genealogy
of a smaller subset of the original samples. This can be done using the powerful
TreeSequence.simplify() method.
The TreeSequence.draw_svg() method allows us to draw
more than one tree: either the entire tree sequence, or
(by using the x_lim parameter) a smaller region of the genome:
reduced_ts = ts.simplify([0, 1, 2, 3, 4, 5, 6, 7, 8, 9]) # simplify to the first 10 samples
reduced_ts.draw_svg(
x_lim=(0, 10000),
title="Genealogy of the first 10 samples for the first 10kb of the genome",
y_axis=True,
y_ticks=[0, 2000, 4000, 6000, 8000],
node_labels = {u: u for u in reduced_ts.samples()},
)

These are much more standard-looking coalescent trees, with far longer branches higher up in the tree, and therefore many more mutations at higher-frequencies.
Note
In this tutorial we refer to objects, such as sample nodes, by their numerical IDs. These can change after simplification, and it is often more meaningful to work with metadata, such as sample and population names, which can be permanently attached to objects in the tree sequence. Such metadata is often incorporated automatically by the tools generating the tree sequence.
Processing sites and mutations#
For many purposes it may be better to focus on the genealogy of your samples, rather than
the sites and
mutations that
define the genome sequence itself. Nevertheless,
tskit also provides efficient ways to return Site and
Mutation objects from a tree sequence.
For instance, under the finite sites model of mutation that we used above, multiple mutations
can occur at some sites, and we can identify them by iterating over the sites using the
TreeSequence.sites() method:
import numpy as np
num_muts = np.zeros(ts.num_sites, dtype=int)
for site in ts.sites():
num_muts[site.id] = len(site.mutations) # site.mutations is a list of mutations at the site
# Print out some info about mutations per site
for nmuts, count in enumerate(np.bincount(num_muts)):
info = f"{count} sites"
if nmuts > 1:
info += f", with IDs {np.where(num_muts==nmuts)[0]},"
print(info, f"have {nmuts} mutation" + ("s" if nmuts != 1 else ""))
0 sites have 0 mutations
6368 sites have 1 mutation
3 sites, with IDs [1355 3767 3806], have 2 mutations
Extracting genotypes#
At each site, the sample nodes will have a particular allelic state (or be flagged as
Missing data). The
TreeSequence.variants() method gives access to
full variation data - allelic states and node genotypes at each site.
Since nodes are by definition haploid, their genotype at a site is simply the
allelic state, but for efficiency, the genotypes attribute
at a site returns a numpy array of integers.
import numpy as np
np.set_printoptions(linewidth=200) # print genotypes on a single line
print("Genotypes")
for v in ts.variants():
print(f"Site {v.site.id}: {v.genotypes}")
if v.site.id >= 4: # only print up to site ID 4
print("...")
break
Genotypes
Site 0: [0 1 0 1 0 1 0 1 1 1 0 0 0 0 1 1 1 1 0 0 0 1 1 1 0 0 1 0 0 1 1 1 1 0 0 1 1 1 0 1]
Site 1: [0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0]
Site 2: [0 1 0 1 0 1 0 1 1 1 0 0 0 0 1 1 1 1 0 0 0 1 1 1 0 0 1 0 0 1 1 1 1 0 0 1 1 1 0 1]
Site 3: [0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 1 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0]
Site 4: [0 0 1 0 1 0 0 0 0 0 1 0 1 1 0 0 0 0 1 1 0 0 0 0 1 0 0 1 0 0 0 0 0 0 1 0 0 0 0 0]
...
Note
Tree sequences are optimised to look at all samples at one site, then all samples at an
adjacent site, and so on along the genome. It is much less efficient to look at all the
sites for a single sample, then all the sites for the next sample, etc. In other words,
you should generally iterate over sites, not samples. Nevertheless, all the alleles for
a single sample can be obtained via the inefficient TreeSequence.haplotypes() method.
If a reference sequence is defined,
To find the actual allelic states at a site, you can refer to the
alleles provided for each Variant:
the genotype value is an index into this list. Here’s one way to print them out; for
clarity this example also prints out the IDs of both the sample nodes (i.e. the genomes)
and the diploid individuals in which each sample
node resides.
samp_ids = ts.samples()
print(" ID of diploid individual: ", " ".join([f"{ts.node(s).individual:3}" for s in samp_ids]))
print(" ID of (sample) node: ", " ".join([f"{s:3}" for s in samp_ids]))
for v in ts.variants():
site = v.site
alleles = np.array(v.alleles) # NB, a shortcut is to use v.states()
print(f"Site {site.id} (ancestral state '{site.ancestral_state}')", alleles[v.genotypes])
if site.id >= 4: # only print up to site ID 4
print("...")
break
ID of diploid individual: 0 0 1 1 2 2 3 3 4 4 5 5 6 6 7 7 8 8 9 9 10 10 11 11 12 12 13 13 14 14 15 15 16 16 17 17 18 18 19 19
ID of (sample) node: 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39
Site 0 (ancestral state 'T') ['T' 'A' 'T' 'A' 'T' 'A' 'T' 'A' 'A' 'A' 'T' 'T' 'T' 'T' 'A' 'A' 'A' 'A' 'T' 'T' 'T' 'A' 'A' 'A' 'T' 'T' 'A' 'T' 'T' 'A' 'A' 'A' 'A' 'T' 'T' 'A' 'A' 'A' 'T' 'A']
Site 1 (ancestral state 'T') ['T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'T' 'A' 'T' 'T' 'T' 'T' 'T']
Site 2 (ancestral state 'T') ['T' 'C' 'T' 'C' 'T' 'C' 'T' 'C' 'C' 'C' 'T' 'T' 'T' 'T' 'C' 'C' 'C' 'C' 'T' 'T' 'T' 'C' 'C' 'C' 'T' 'T' 'C' 'T' 'T' 'C' 'C' 'C' 'C' 'T' 'T' 'C' 'C' 'C' 'T' 'C']
Site 3 (ancestral state 'C') ['C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'G' 'C' 'C' 'C' 'C' 'C' 'G' 'G' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C' 'C']
Site 4 (ancestral state 'C') ['C' 'C' 'G' 'C' 'G' 'C' 'C' 'C' 'C' 'C' 'G' 'C' 'G' 'G' 'C' 'C' 'C' 'C' 'G' 'G' 'C' 'C' 'C' 'C' 'G' 'C' 'C' 'G' 'C' 'C' 'C' 'C' 'C' 'C' 'G' 'C' 'C' 'C' 'C' 'C']
...
Note
Since we have used the msprime.JC69 model of mutations, the alleles are all
either ‘A’, ‘T’, ‘G’, or ‘C’. However, more complex mutation models can involve mutations
such as indels, leading to allelic states which need not be one of these 4 letters, nor
even be a single letter.
Computing statistics#
There are a large number of statistics and related calculations
built in to tskit. Indeed, many basic population genetic statistics are based
on the allele (or site) frequency spectrum (AFS), which can be obtained from a tree sequence
using the TreeSequence.allele_frequency_spectrum()
method:
afs = ts.allele_frequency_spectrum()
plt.bar(np.arange(ts.num_samples + 1), afs)
plt.title("Unpolarised allele frequency spectrum")
plt.show()
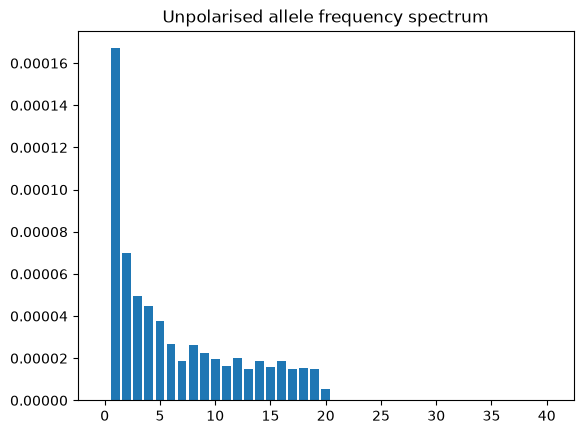
By default this method returns the “folded” or unpolarized AFS that doesn’t take account of the ancestral state. However, since the tree sequence provides the ancestral state, we can plot the polarized version; additionally we can base our calculations on branch lengths rather than alleles, which provides an estimate that is not influenced by random mutational “noise”.
fig, (ax1, ax2) = plt.subplots(ncols=2, figsize=(12, 3))
afs1 = ts.allele_frequency_spectrum(polarised=True, mode="branch")
ax1.bar(np.arange(ts.num_samples+1), afs1)
ax1.set_title("Genome-wide branch-length AFS")
restricted_ts = ts.keep_intervals([[5e6, 5.5e6]])
afs2 = restricted_ts.allele_frequency_spectrum(polarised=True, mode="branch")
ax2.bar(np.arange(restricted_ts.num_samples+1), afs2)
ax2.set_title("Branch-length AFS between 5 and 5.5Mb")
plt.show()
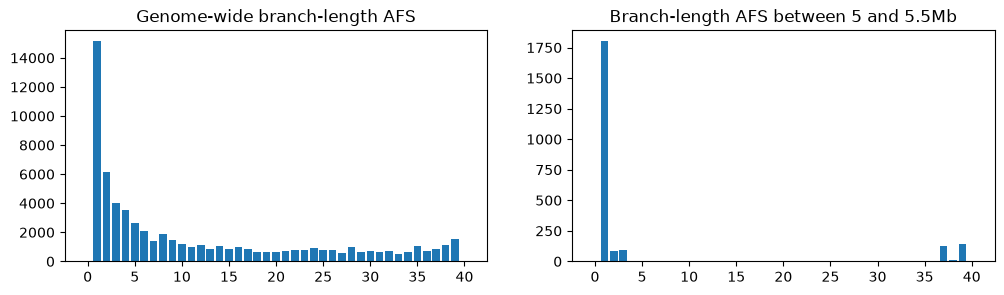
On the left is the frequency spectrum averaged over the entire genome, and on the right
is the spectrum for a section of the tree sequence between 5 and 5.5Mb, which we’ve
created by deleting the regions outside that interval using
TreeSequence.keep_intervals(). Unsurprisingly,
as we noted when looking at the trees, there’s a far higher proportion of singletons in
the region of the sweep compared to the entire genome.
Windowing#
It is often useful to see how statistics vary in different genomic regions. This is done
by calculating them in windows along the genome. For this,
let’s look at a single statistic, the genetic diversity() (π). As a
site statistic, it measures the average number of genetic differences between two
randomly chosen samples, whereas as a branch statistic, it measures the average
branch length between them. We’ll plot how the value of π changes using 10kb windows,
plotting the resulting diversity between positions 4 and 6 Mb:
fig, (ax1, ax2) = plt.subplots(ncols=2, figsize=(12, 3))
L = int(ts.sequence_length)
windows = np.linspace(0, L, num=L//10_000 + 1)
ax1.stairs(ts.diversity(windows=windows), windows/1_000, baseline=None) # Default is mode="site"
ax1.set_ylabel("Diversity")
ax1.set_xlabel("Genome position (kb)")
ax1.set_title("Site-based calculation")
ax1.set_xlim(4e3, 6e3)
ax1.set_yscale("log")
ax1.set_ylim(1e-6, 1e-3)
ax2.stairs(ts.diversity(windows=windows, mode="branch"), windows/1_000, baseline=None)
ax2.set_xlabel("Genome position (kb)")
ax2.set_title("Branch-based calculation")
ax2.set_xlim(4e3, 6e3)
ax2.set_yscale("log")
plt.show()
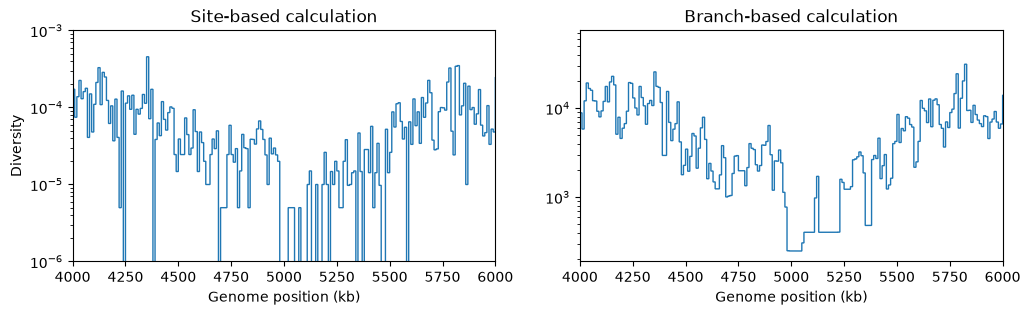
There’s a clear drop in diversity in the region of the selective sweep. And as expected, the statistic based on branch lengths gives a less noisy signal than the statistic based on randomly occurring mutations at sites.
Saving and exporting data#
Tree sequences can be efficiently saved to file using TreeSequence.dump(), and
loaded back again using tskit.load(). By convention, we use the suffix .trees
for such files:
import tskit
ts.dump("data/my_tree_sequence.trees")
new_ts = tskit.load("data/my_tree_sequence.trees")
It’s also possible to export tree sequences to different formats. Note, however, that not only are these usually much larger files, but that analysis is usually much faster when performed by built-in tskit functions than by exporting and using alternative software. If you have a large tree sequence, you should try to avoid exporting to other formats.
Newick and Nexus format#
The most common format for interchanging tree data is Newick. We can export to a newick format string quite easily. This can be useful for interoperating with existing tree processing libraries but is very inefficient for large trees. There is also no support for including sites and mutations in the trees.
small_ts = reduced_ts.keep_intervals([[0, 10000]])
tree = small_ts.first()
print(tree.newick(precision=3))
(((3:46.300,5:46.300):2121.638,(1:125.460,7:125.460):2042.478):6071.237,(4:6868.532,((6:729.815,(8:440.016,9:440.016):289.799):1102.530,(2:1333.615,10:1333.615):498.730):5036.186):1370.642);
For an entire set of trees, you can use the Nexus file format, which acts as a container for a list of Newick format trees, one per line:
small_ts = small_ts.trim() # Must trim off the blank region at the end of cut-down ts
print(small_ts.as_nexus(precision=3, include_alignments=False))
#NEXUS
BEGIN TAXA;
DIMENSIONS NTAX=10;
TAXLABELS n0 n1 n2 n3 n4 n5 n6 n7 n8 n9;
END;
BEGIN TREES;
TREE t0.000^613.000 = [&R] (((n2:46.300,n4:46.300):2121.638,(n0:125.460,n6:125.460):2042.478):6071.237,(n3:6868.532,((n5:729.815,(n7:440.016,n8:440.016):289.799):1102.530,(n1:1333.615,n9:1333.615):498.730):5036.186):1370.642);
TREE t613.000^5479.000 = [&R] (((n2:46.300,n4:46.300):2121.638,(n0:125.460,n6:125.460):2042.478):6071.237,(n3:1935.187,((n5:729.815,(n7:440.016,n8:440.016):289.799):1102.530,(n1:1333.615,n9:1333.615):498.730):102.842):6303.987);
TREE t5479.000^10000.000 = [&R] ((n2:46.300,n4:46.300):8192.874,((n0:125.460,n6:125.460):3081.030,(n3:1935.187,((n5:729.815,(n7:440.016,n8:440.016):289.799):1102.530,(n1:1333.615,n9:1333.615):498.730):102.842):1271.303):5032.684);
END;
VCF#
The standard way of interchanging genetic variation data is the Variant Call Format (VCF), for which tskit has basic support:
import sys
small_ts.write_vcf(sys.stdout)
##fileformat=VCFv4.2
##source=tskit 1.0.3
##FILTER=<ID=PASS,Description="All filters passed">
##contig=<ID=1,length=10000>
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT tsk_0 tsk_1 tsk_2 tsk_3 tsk_4
1 3002 0 T A . PASS . GT 0|1 0|1 0|1 0|1 1|1
1 4427 1 T C . PASS . GT 0|1 0|1 0|1 0|1 1|1
1 5770 2 C G . PASS . GT 0|0 1|0 1|0 0|0 0|0
1 6509 3 T A . PASS . GT 0|0 0|0 0|0 0|0 0|1
The TreeSequence.write_vcf() method takes a file object as a parameter;
to get it to write out to the notebook here we ask it to write to stdout.
Scikit-allel#
Because tskit integrates very closely with numpy, we can interoperate very efficiently with downstream Python libraries for working with genetic sequence data, such as scikit-allel. We can interoperate with scikit-allel by exporting the genotype matrix as a numpy array, which scikit-allel can then process in various ways.
import allel
# Export the genotype data to allel. Unfortunately there's a slight mismatch in the
# terminology here where genotypes and haplotypes mean different things in the two
# libraries.
h = allel.HaplotypeArray(small_ts.genotype_matrix())
print(h.n_variants, h.n_haplotypes)
h
4 10
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 1 |
| 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 1 |
| 2 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
scikit-allel has a wide-ranging and efficient suite of tools for working with genotype data for most users’ needs. For example, it gives us an another way to compute the pairwise diversity statistic:
ac = h.count_alleles()
allel.mean_pairwise_difference(ac)
array([0.53333333, 0.53333333, 0.35555556, 0.2 ])
Here’s the equivalent using the native TreeSequence.diversity() method,
windowed by site:
# Per site (not span-normalised to match scikit-allel)
small_ts.diversity(windows="sites", span_normalise=False)
array([0.53333333, 0.53333333, 0.35555556, 0.2 ])
Note, however, that you would normally want to set span_normalise=True to account
for the fact that sites are not evenly spread along the genome (see these docs).
Key points covered above#
Here are the simple methods and take-home messages from this introduction to the tskit Python API, in rough order of importance:
Objects and their attributes
In Python, a
TreeSequenceobject has a number of basic attributes such asnum_trees,num_sites,num_samples,sequence_length, etc. Similarly aTreeobject has e.g. anintervalattribute, aSiteobject has amutationsattribute, aNodeobject has atimeattribute, and so on.Nodes (i.e. genomes) can belong to individuals. For example, sampling a diploid individual results in an
Individualobject which links to two distinct sample nodes.
Key tree sequence methods
samples()returns an array of node IDs specifying the nodes that are marked as samplesnode()returns the node object for a given integer node IDtrees()iterates over all the treessites()iterates over all the sitesvariants()iterates over all the sites with their list of unique alleles and genotypessimplify()reduces the number of sample nodes in the tree sequence to a specified subsetkeep_intervals()(or its complement,delete_intervals()) removes genetic information from specific regions of the genomedraw_svg()returns an SVG representation of a tree sequence (and plots it if in a Jupyter notebook). Similarly,Tree.draw_svg()plots individual trees.at()returns a tree at a particular genomic position (but usingtrees()is usually preferable)Various population genetic statistics can be calculated using methods on a tree sequence, for example
allele_frequency_spectrum(),diversity(), andFst(); these can also be calculated in windows along the genome.