What's new in tskit 0.6.0!
The 0.6.0 release of tskit is a major release with significant enhancements to genetic analysis capabilities, tree visualization, and general usability.
Breaking changes
The definition of ts.genetic_relatedness()
and ts.genetic_relatedness_weighted()
are changed to average over sample sets.
This allows comparison between sample sets of different sizes. For example this will change the result
by a factor of four for diploid sample sets. The default for these methods is also changed to
polarised=True, but the output is unchanged for centre=True (the default). Thanks to
@petrelharp and @mmosmond for their work on this.
New features
Pair_coalescence functions
Thanks to @nspope, tskit now has a number of new methods for
calculating coalescence events and rates.
The ts.pair_coalescence_counts()
method calculates coalescence events per node or time interval, and can be used to calculate
the CDF of coalescences over time. The inverse of the empirical CDF, allowing quantiles to be
calculated, is given by
ts.pair_coalescence_quantiles(). Finally, ts.pair_coalescence_rates()
estimates instantaneous rates of pair coalescence within time intervals.
These methods can be passed both time_windows, and windows that correspond to genomic
intervals, allowing pairwise coalescence metrics to be calculated over both time and
genomic space.
# Make a model with a selective sweep in the middle of a 10Mb genome
>>> L = 10e6 # 10 Mb
>>> params = {"sequence_length": L, "population_size":1e4, "recombination_rate": 1e-8}
>>> sweep_model = msprime.SweepGenicSelection(
... position=L/2, start_frequency=0.0001, end_frequency=0.9999, s=0.25, dt=1e-6)
>>> ts = msprime.sim_ancestry(20, model=[sweep_model, msprime.StandardCoalescent()], **params)
>>> time_intervals = np.logspace(0, np.log10(ts.max_time), 20)
# time intervals need to go from 0 -> infinity
>>> time_intervals = np.concatenate(([0], time_intervals, [np.inf]))
>>> genome_intervals = np.linspace(0, L, 21)
>>> counts = ts.pair_coalescence_counts(time_windows=time_intervals, windows=genome_intervals)
>>> print(np.array_repr(counts, max_line_width=100, precision=6, suppress_small=True))
array([[ 0. , 0. , 0. , ..., 35.51717 , 4.875602, 0. ],
[ 0. , 0. , 0. , ..., 17.337302, 0.167958, 0. ],
[ 0. , 0. , 0. , ..., 14.809552, 0. , 0. ],
...,
[ 0. , 0. , 1. , ..., 17.95416 , 0.00702 , 0. ],
[ 0. , 0. , 1. , ..., 21.790546, 0.645824, 0. ],
[ 0. , 0. , 1. , ..., 33.664214, 0. , 0. ]])
Here the number of coalescences in each block of time and genomic region have been normalised
by dividing by the span of each window (you can specify span_normalise=False to change this).
As the time windows are logarithmically spaced, to normalise by the size of the time window, use
pair_coalescence_rates.
In the example above, this clearly highlights the effect of selection at a particular point
in the genome at a particular time:
>>> import seaborn as sns
>>> rates = ts.pair_coalescence_rates(time_windows=time_intervals, windows=genome_intervals)
>>> ax = sns.heatmap(rates.T, vmin=0, vmax=np.max(rates[np.isfinite(rates)]), cbar_kws={'label': 'Coalescence rate'})
>>> ax.set_xticks(np.arange(len(genome_intervals) - 0.5), genome_intervals/1e6, rotation=90)
>>> ax.set_yticks(np.arange(len(time_intervals) - 0.5), [f"{t:.2f}" for t in time_intervals])
>>> ax.invert_yaxis()
>>> ax.set_xlabel("Genome position")
>>> ax.set_ylabel("Time");
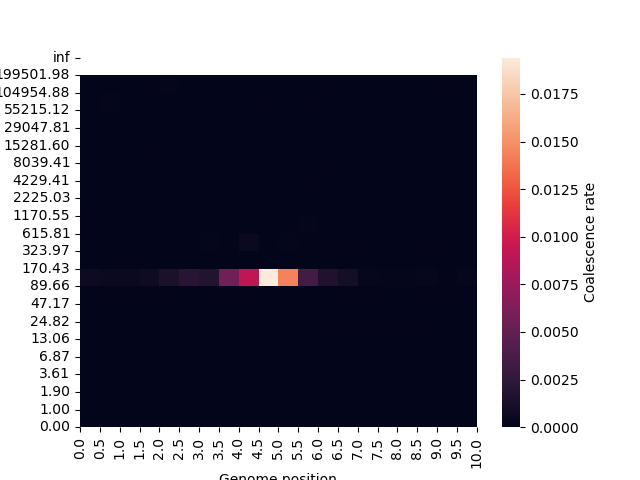
Specifying different sample sets to these functions allows cross-coalescence metrics to be calculated, which helps to reveal historical patterns of mixing between different populations.
Extending haplotypes
@petrelharp, @hfr1tz3, @nspope, and @avabamf have put together a new method ts.extend_haplotypes that reduces the number of edges in a tree sequence by extending ancestral haplotypes. This makes the tree sequence smaller, while adding unary nodes.
>>> ts = msprime.sim_ancestry(10000, recombination_rate=0.001, sequence_length=10000)
>>> ts.num_edges
41512
>>> ts.extend_haplotypes().num_edges
40761
See this previous news item for a summary of our recent paper discussing the importance of unary nodes in tree sequences.
New Tree methods
Several new methods have been added to the Tree class. @Billyzhang1229 has added both a
distance_between
method that calculates the total distance between two nodes in a tree, essentially adding the branch lengths separating them,
and an rf_distance method
that calculates the unweighted Robinson-Foulds distance between two trees.
@hyanwong has added an
ancestors method that returns the ancestors of a given node.
# Calculate the total distance between two nodes
>>> ts.at(547630.5).distance_between(42,43)
5578.6766720892065
# Calculate the Robinson-Foulds distance between two trees
>>> ts.at(547630.5).rf_distance(ts.at(647630))
192
# Get the ancestors of a node as an iterator
>>> list(ts.at(547630.5).ancestors(42))
[606, 931, 1863, 2483, 2570, 2590, 3603, 3913]
Additional helper properties and methods
Several quality-of-life improvements have been added. Edges now have an interval attribute that returns a tskit.Interval object thanks to @hyanwong and those Intervals and Trees now have mid properties that return the midpoint of the interval or tree. Thanks to @currocam for this addition. Dropping metadata from a table is now simpler with the new Table.drop_metadata method, thanks to @jeromekelleher.
# Get the interval of an edge
>>> ts.edge(42).interval
Interval(left=194621.0, right=900640.0)
# Get the midpoint of an interval
>>> ts.edge(42).interval.mid
547630.5
# Drop metadata from a table
table.drop_metadata()
@hyanwong has also added a
states() method to Variant objects.
This returns the genotypes as an array of strings, rather than integer indexes, to aid comparison of genetic variation.
Note that this is an inefficient way to access the genotypes, but can be useful for showing the actual alleleic states.
>>> next(ts.variants()).states()
array(['G', 'G', 'G', 'C', 'G', 'G', 'G', 'G', 'G', 'G', 'G', 'G', 'G',
'G', 'G', 'G', 'G', 'G', 'G', 'G', 'G', 'G', 'G', 'G', 'G', 'G',
'G', 'C',], dtype='<U1')
Genetic relatedness
@jeromekelleher and @petrelharp have added a ts.genetic_relatedness_matrix() method that computes the full pairwise genetic relatedness between and within pairs of sets of nodes from sample sets, and a ts.genetic_relatedness_vector() method that computes the product of the genetic relatedness matrix and a weight vector.
>>> ts.genetic_relatedness_matrix(sample_sets=((0,1),(2,3)))
array([[ 2.37625e-05, -2.37625e-05],
[-2.37625e-05, 2.37625e-05]])
More is planned for genetic relatedness metrics in future tskit versions, which should allow e.g. efficient calculation of PCAs.
Drawing improvements
Finally, the draw_svg()
methods now have a number of new features, via @hyanwong.
Gene annotations can now be added to the x-axis using the x_regions parameter, and a titles can be
placed at the top of the drawing. When viewed in HTML, it is possible to specify text to appear when
mousing over nodes or mutations using the node_titles and mutation_titles parameters:
>>> from IPython.display import HTML
>>> ts = msprime.sim_ancestry(2, sequence_length=1000, recombination_rate=0.002, random_seed=123)
>>> genic_regions = {(10, 100): "Gene 1", (700, 950): "Gene 2"}
>>> svg = ts.draw_svg(
... x_axis=True, size=(800, 300), max_num_trees=4, x_regions=genic_regions,
... title="SVG tree sequence plots can now have titles!",
... # Hide the 3rd X tick labels, which clash with the gene regions
... style=".x-axis .tick:nth-child(2) .lab, .x-axis .tick:nth-child(4) .lab {display: none}",
... node_titles={u: f"This is sample node {u}" for u in ts.samples()},
... )
>>> HTML(svg) # For mouseover tooltips to appear in a Jupyter notebook, display the plot as html
The square sample nodes of the SVG drawing above should display additional text on mouseover
Experimental drawing options
For testing purposes, we have introduced experimental features which can help draw large trees.
However, the API for this is not finalised, hence the feature is currently undocumented: feedback is welcome.
In particular the order parameter of the
Tree.draw_svg() method
can now also take a (postorder) subset of nodes, allowing clades to be visually collapsed, and
new pack_untracked_polytomies parameter allows large polytomies involving untracked samples to be
summarised as a dotted line:
 |
 |